
PAH disease progression
Pulmonary arterial hypertension (PAH) is a rare and progressive disease that primarily affects the lung vasculature and small pulmonary arterioles.1,2 Normal pulmonary circulation occurs through a low-pressure, high-flow system that includes the lungs and heart.2 In PAH, progressive remodeling and the obliteration of the pulmonary arterioles cause increased pressures in the lungs.2 Although at first the heart adapts to pressure changes, the rising pulmonary pressures eventually lead to right heart ventricular dysfunction, heart failure, and death.3-5
Lung and heart changes during PAH progression
Lung progression
Normal

Pulmonary arteries make up an extensive network in the lungs4
Pulmonary arteries have a healthy endothelium, allowing for normal perfusion4
Compensation

As PAH progresses, remodeling of the small to medium pulmonary arteries causes them to become narrow and stiff, restricting blood flow2,5
Microvessel loss leads to a mild increase in PVR and a moderate decrease in perfusion4
Failure

Cell proliferation and obliterative remodeling in the pulmonary arteries lead to a severe increase in PVR and a severe decrease in perfusion4
Right heart progression
Normal
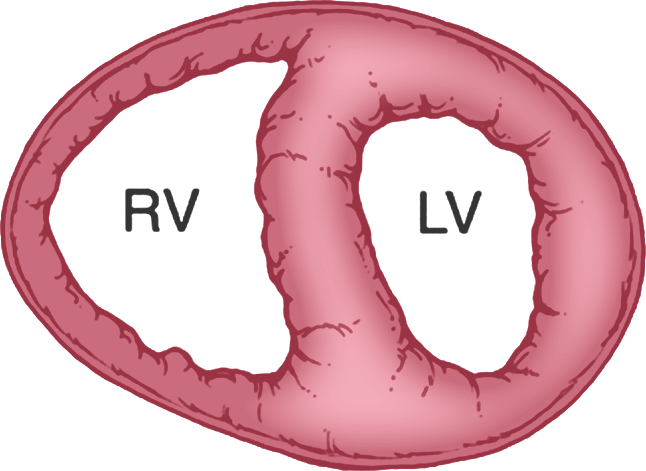
RV is thin-walled and has a normal CO due to healthy pulmonary circulation4
Compensation
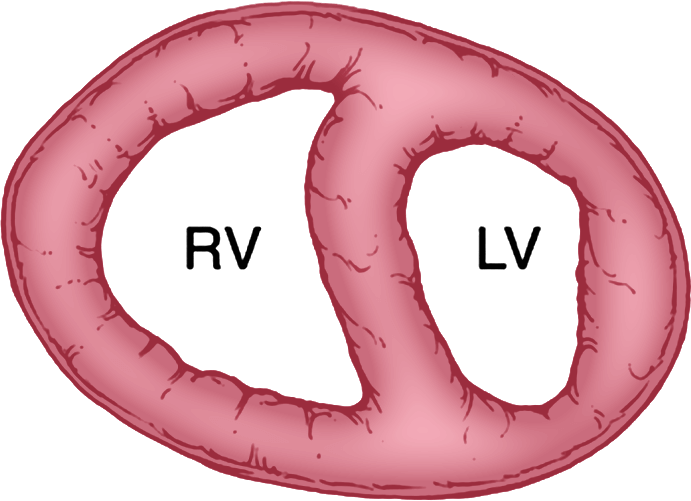
RV hypertrophies and CO remains preserved
At this stage, there are no additional patient symptoms. Hemodynamics are only minimally affected, if at all4
Failure
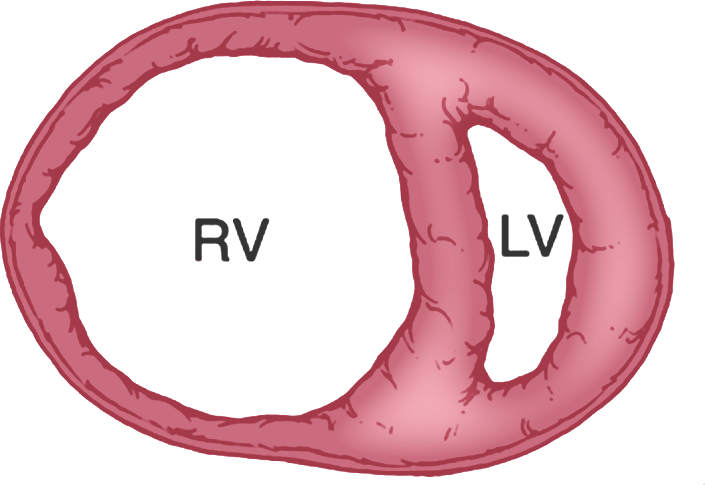
Over time, the RV dilates, and CO is severely depressed, leading to right heart failure4,5
RV dysfunction in PAH
Although PAH is a disease of the lungs, it severely affects the right side of the heart―both anatomically and functionally―ultimately leading to right heart failure.2,5
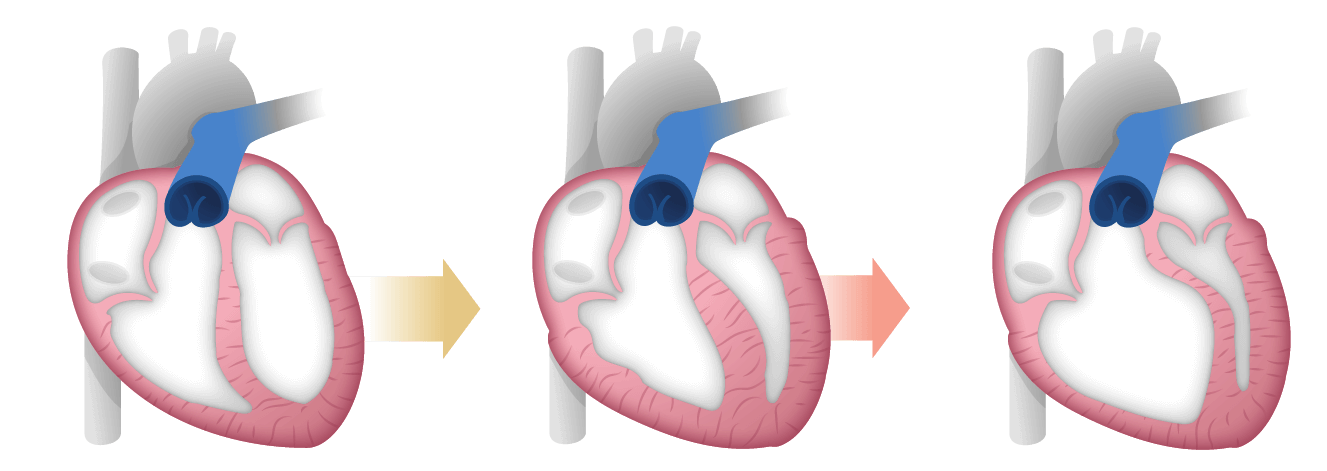
Preservation of CO is critical in PAH6

PAH raises PVR and mPAP, placing strain on the RV2,4,5,7:
- Narrowing of pulmonary arterioles increases PVR and mPAP2
- RV initially adapts by working harder and remodeling to maintain CO, which delays symptoms4
- This compensation is limited; as PVR continues to rise, CO decreases, especially during exertion7
- Persistent high pressures lead to RV hypertrophy, decompensation, and reduced lung and systemic blood flow4
- RV failure results in elevated RAP and decreased mPAP7
- The progression ultimately leads to right heart failure, the primary cause of death in PAH1,2,8
Echo imaging reveals right heart dilation
Echo provides essential visualization of right heart changes, aiding in detecting right heart dilation and dysfunction as the disease progresses.4,9
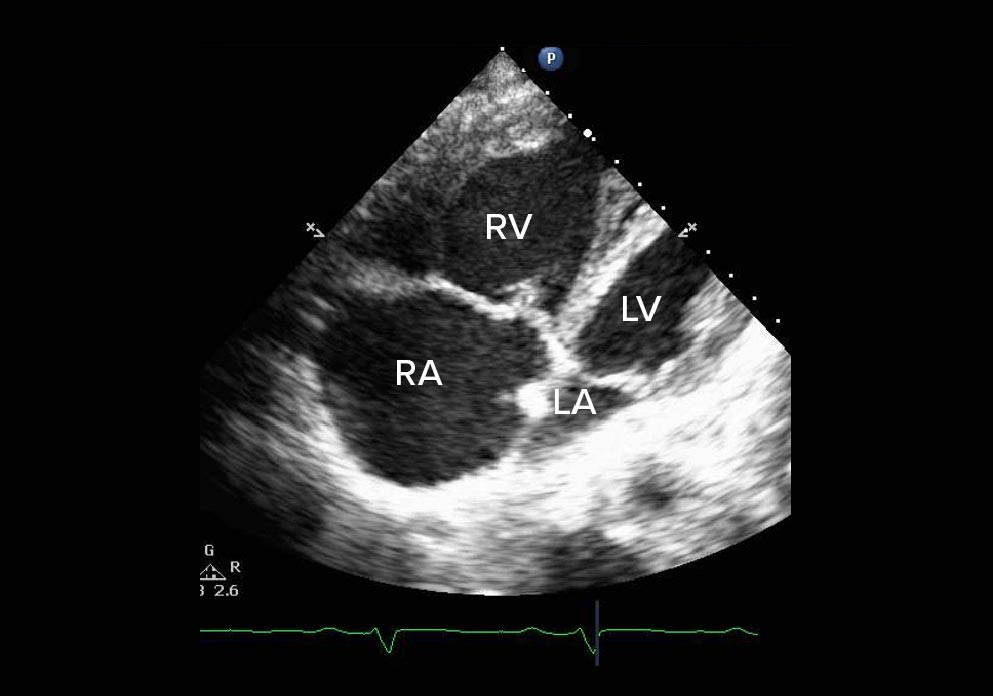
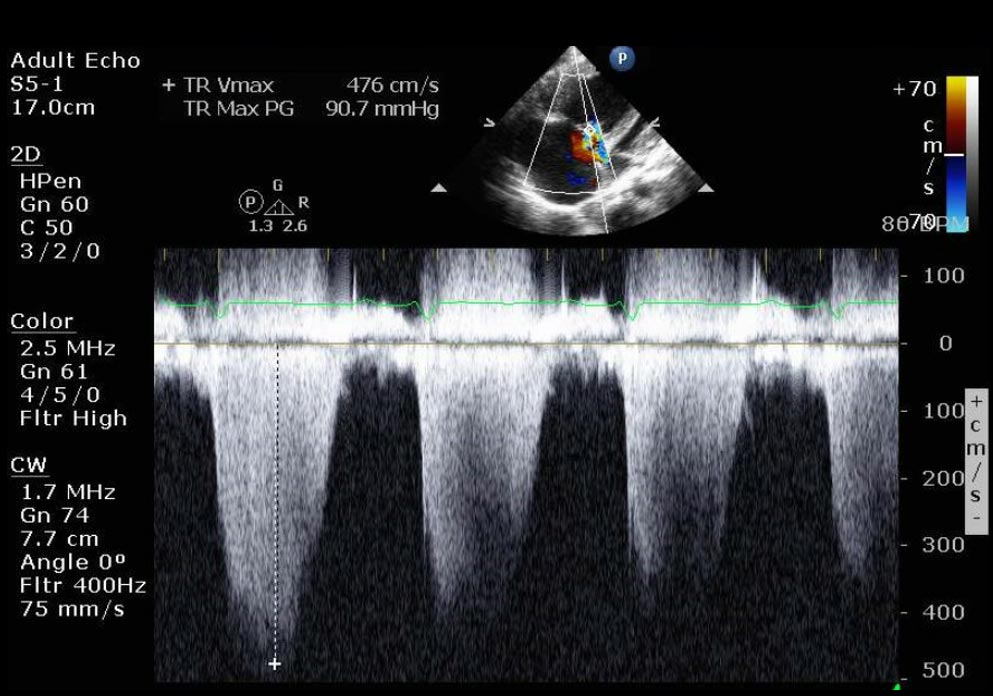
Apical 4-chamber view
- Paradoxical wall motion septum
- Severe RV and RA dilation
Doppler: Systolic pulmonary artery pressure
- RA pressure estimated to be 15 mm Hg due to noncompressible dilated inferior vena cava
- Systolic pulmonary artery pressure: 96 mm Hg
Addressing the pathophysiology of PAH
When monitoring your patient’s response to treatment, remember that PAH is a progressive disease—even if outward symptoms are not evident. Proactive monitoring of the right heart is critical, as detrimental changes to the RV typically occur before declines are seen in a patient’s FC or 6MWD.10,11